|
Research Areas
Our group uses a combination of theory, computer simulation, and experiment to study structure, dynamics, and function of proteins. The research follows three general directions: structure, thermodynamics, and kinetics of protein association; functional mechanisms of ion channels; and crowding and confinement effects of proteins in cellular environments.
Protein Association: Structure, Thermodynamics, and Kinetics
Using knowledge accumulated from the interfaces of known protein-protein and protein-DNA complexes, we have developed neural-network methods, cons-PPISP and DISPLAR, for predicting interfaces residues. The methods promise to complement experimental methods such as chemical-shift perturbations for characterizing interfaces. Among other applications, these methods are used to generate data for driving docking and for scoring docked structures. This use of cons-PPISP has found successes in recent rounds of CAPRI ("Critical Assessment of PRediction of Interactions").
While hydrophobic interactions are well established as a driving force for protein folding and binding, the contributions of electrostatic interactions have been controversial. A widely held view in the theoretical community is that electrostatic interactions either have marginal effects or are destabilizing for structure formation and assembly of proteins and nucleic acids. This view originates from the fact that, according to Poisson-Boltzmann calculations, electric charges pay a significant desolvation cost upon protein folding or binding. In a series of studies, our group showed that the magnitude of the desolvation cost is very sensitive to model parameters (in particular how the boundary between the protein or nucleic acid molecule and the solvent is defined). The conventional choice for the dielectric boundary is the molecular surface. We found that using the van der Waals surface as the dielectric boundary consistently gives better agreement with experimental results for mutational effects on folding and binding stability, and leads to the observed electrostatic enhancement of many protein association rates. The van der Waals surface also appears more physical in that the presence of high-dielectric crevices mimicks the the penetration of water into protein interior. Our studies lead to the conclusion that charged groups on proteins and nucleic acids generally can significantly stabilize folded structures and assemblies. We have also made considerable strides in speeding up electrostatic calculations through the development of a generalized Born method, GBr6.
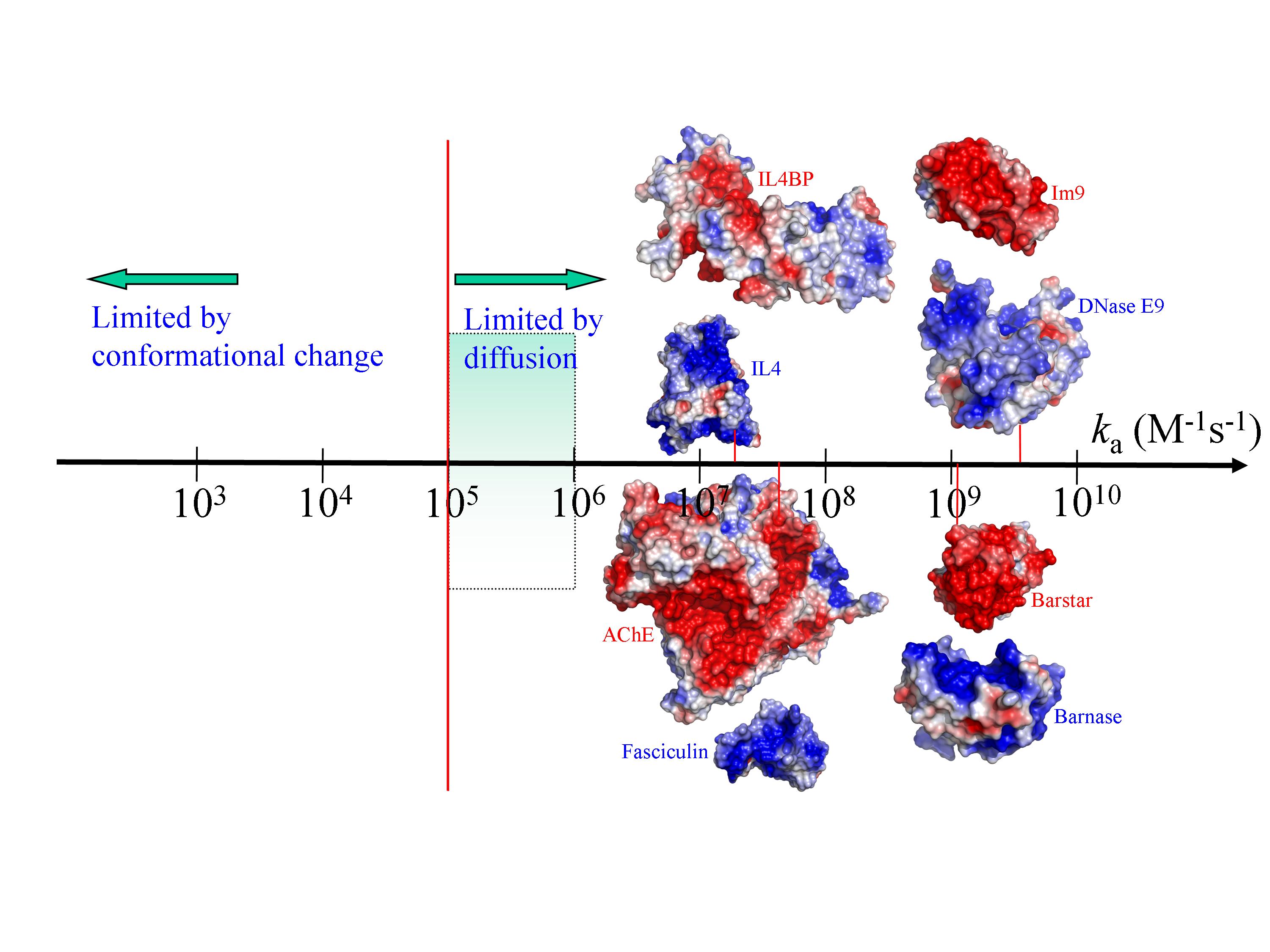
A long-term interest of our group is the rates of protein association. Association rates have received heightened attention in recent years, in particular constituting a main ingredient of systems biology. Proteins are generally a part of interaction networks and the underlying biological processes may often be under kinetic control rather than thermodynamic control. Our research over the years culminated in the transient-complex theory for protein association, which presents a computational methodology for elucidating the mechanisms of protein association and for predicting the association rates. This theory provides a simple rationalization of the variations in observed association rates over 10 orders of magnitudes and for accurate prediction of association rates based on the structures of the native complexes. The methodology is now available through the TransComp web server.
Functional Mechanisms of Ion Channels

Dynamics is now recognized as the critical link between structure and function. In a recent study, we captured spontaneous conformational transition to the open state of the acetylcholine receptor in the unliganded form by molecular dynamics simulations. The acetylcholine receptor is a prototype for ligand-gated ion channels. Our study suggests that the unliganded form, although predominantly sampling the closed state, can make excursions into the open state. The open state has high affinity for agonists, leading to channel activation, whereas the closed state upon distortion has high affinity for antagonists, leading to inhibition.
Based on molecular dynamics simulations, we have also developed models for the funcional mechanisms of the glutamate receptor (another ligand-gated ion channel) and the M2 proton channel of influenza virus A.
Crowding and Confinement in Cellular Environments
How relevant are the results of in vitro experiments, typically obtained in dilute solutions, to biochemical processes in crowded and compartmentalized environments of cells? We have developed statistical-thermodyanmic theories to obtain qualitative estimates of crowding and confinement effects on protein folding and binding thermodynamics and kinetics. These estimates are largely confirmed by recent experiments, including those carried out by ourselves. We are tackling new challenges, such as the modeling of atomic details of crowding molecules and confining environments, and the study of crowding effects on cell membranes.
|